Синдром Марфана
Синдром Марфана – наследственная патология соединительной ткани, проявляющаяся отклонениями в строении и работе многих систем организма. Задействованы костно-суставная, зрительная, сердечно-сосудистая, дыхательная системы, кожа.
Встречается у 1 человека на 10 000 населения. Степень выраженности нарушений сильно варьируется: от незначительных, не доставляющих дискомфорта, или случайных находок при проведении дополнительных методов исследований до тяжёлых, приводящих к инвалидности и ранней смерти. Средняя продолжительность жизни таких больных 30-40 лет. В 90% случаев к смерти приводят осложнения со стороны сердца или крупных сосудов: разрыв аневризмы аорты, септический эндокардит.
Содержание
Критерии диагностики
Описано заболевание было в 1896 году во Франции педиатром Марфаном. В 1986 году были установлены чёткие критерии заболевания. Доказано, что синдром Марфана связан с мутацией генов, отвечающих за синтез белков соединительной ткани: фибриллина 1 и 2. Однако есть много других наследственных болезней с тем же дефектом генов и похожим внешним видом. Поэтому только одновременное наличие у больного молекулярных нарушений, подтверждённых исследованиями, и клинических проявлений даёт основание для постановки данного диагноза.
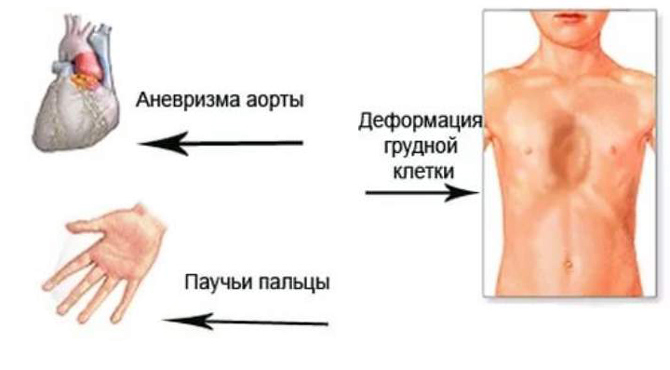
Костная система
Внешний вид больного уже может натолкнуть на мысль о заболевании Марфана. Характерно увеличение длины конечностей и пальцев, удлинённая форма черепа, готическое (высокое) нёбо. Телосложение обычно худощавое, высокий рост, часто выявляют искривления позвоночника и грудной клетки. Наиболее значимые признаки, часто встречающиеся при болезни Марфана и редко при другой патологии, – называют большими критериями.
К ним относят:
- Изменение формы грудной клетки: она выступает вперёд, как у птиц (килевидная) или вогнутая в виде воронки. Асимметрия: слева грудино-рёберные соединения выступают кпереди.
- Изменение пропорций тела: высота верхней части тела меньше нормы (рост сидя), общий рост меньше, чем размах рук более, чем на 5%.
- Увеличена длина пальцев: при обхвате запястья 1 и 5 пальцами другой руки остаётся свободное место или последние фаланги накладываются друг на друга («+» тест запястья). Если большой палец расположить поперёк ладони, он выступает за край кисти («+» тест большого пальца). Удлинение конечностей и пальцев видно уже при рождении.
- Боковое искривление позвоночника более 20 градусов, чаще вправо (присутствует у 60% всех больных). Спондилолистез (6%) – ограниченное разгибание позвоночника, подтверждённое рентгенограммой. Может быть сглажен грудной кифоз (патологически ровная спина) или наоборот, усилен.
- Ограничение подвижности локтевых суставов (анкилоз): человек не может разогнуть руку в локте более 170 градусов.
- Выраженное продольное плоскостопие, которое приводит к смещению внутренних лодыжек к середине.
- Протрузия вертлужной впадины, подтверждённая на рентгенограмме, КТ, МРТ: головка бедренной кости вдавливается в полость малого таза вплоть до её проваливания.
Малые признаки без присутствия больших не учитываются, т. к. распространены и при других патологиях: повышенная подвижность суставов, удлинённый череп, аномалии лицевого скелета (глубокое расположение глаз, мало развитые скулы, высокое нёбо).
Для постановки диагноза необходимо 2 больших или 1 большой и 4 малых критерия.
Зрение
Из-за несовершенства соединительной ткани у больных страдает зрение. Характерна миопия (близорукость), у многих косоглазие. Повышенный риск отслойки сетчатки, поэтому противопоказаны большие физические нагрузки, прыжки, подъём тяжестей, групповые виды спорта.
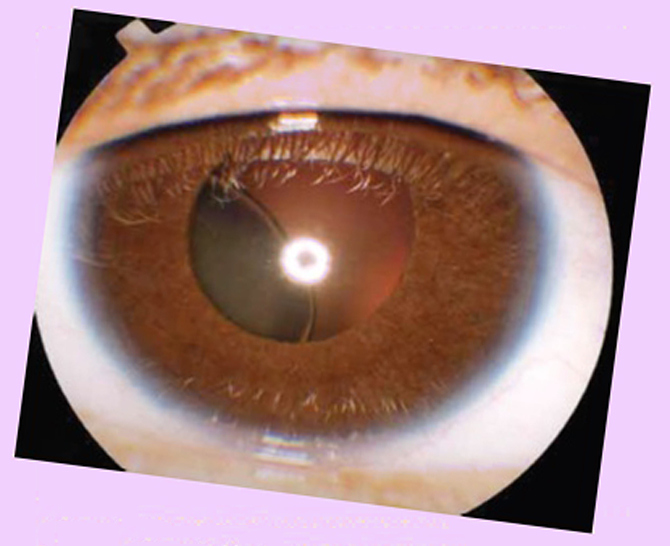
К большим критериям относят эктопию хрусталика. Это смещение хрусталика с одной или обеих сторон в любом направлении, чаще вверх.
Малыми критериями считают:
- увеличение длины глаза, что ведёт к близорукости;
- роговица имеет уплощённую форму;
- недоразвитие радужной оболочки и мышцы, отвечающей за аккомодацию.
1 большой или 2 малых критерия позволяют говорить о вовлечении в патологический процесс зрительной системы. Их диагностирует офтальмолог после проверки остроты зрения, осмотра глазного дна при расширенном зрачке с помощью щелевой лампы, проведения кератометрии (измерения кривизны роговицы) и ультрасонографии (измерения длины оси глазного яблока).
Сердце и сосуды
Патология этой системы часто является определяющей при прогнозе для жизни больного. Причиной смерти у 95% больных в возрасте до 40 лет является аневризма аорты с её осложнениями. Слабость соединительной ткани в стенке сердца и аорты приводит к их расширению, формированию пороков сердца, нарушению нормального тока крови в них (регургитация – обратный ток крови из-за расширения отверстия и неполного смыкания клапанов).
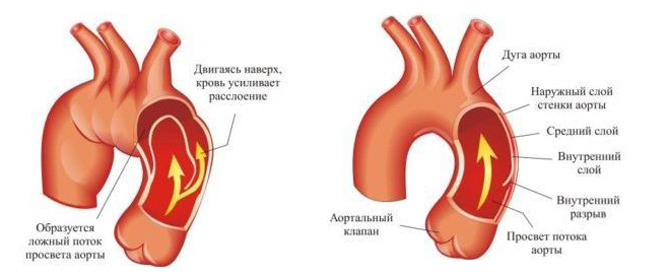
Особенно повышается нагрузка на систему кровообращения во время беременности и родов.
Большими критериями считают:
- расширение восходящего отдела аорты;
- её расслоение.
К малым относят:
- пролапс митрального клапана;
- расширение лёгочного ствола без явных причин на то у людей моложе 40 лет;
- отложение солей кальция в области митрального клапана до 40 лет;
- расширение или расслоение аорты в нисходящем (грудном или брюшном) отделе.
При наличии 1 большого или 1 малого критериев говорят о поражении сердечно-сосудистой системы при синдроме Марфана. Для диагностики проводят ЭхоКГ (замеряют диаметр сосудов, обнаруживают нарушение движений створок клапанов), ангиографию с использованием контраста (подтверждает расслоение аорты), КТ, МРТ органов грудной полости.
Поражение твёрдой мозговой оболочки
Это большой критерий. При его выявлении с большой долей вероятности можно говорить о наличии синдрома Марфана. Он обнаруживается у 40% всех больных. Выявляют при помощи МРТ или КТ пояснично-крестцового отдела позвоночника. На уровне L 5-S 1 (переход поясницы в крестец) спинномозговой канал и отверстия выхода нервных корешков расширены, на телах позвонков истончена замыкательная пластинка, возможна передняя мозговая грыжа (выпячивание твёрдой мозговой оболочки за пределы спинномозгового канала).
Другие симптомы
Из-за слабости соединительнотканных элементов кожи часто образуются полосы втяжения, или стрии. Расположены они, как правило, на плечах, бёдрах, спине. Они похожи на полосы, образующиеся после беременности, резкого похудения, не являются высокоспецифичными для синдрома Марфана, относятся к малым критериям.
Другим возможным проявлением нарушения строения и функции соединительной ткани являются частые грыжи у таких больных. Причём после оперативного лечения они имеют тенденцию к повторению. Именно рецидив грыжи рассматривается как малый признак при синдроме Марфана.
Вовлечение дыхательной системы проявляется образованием поверхностно расположенных булл в лёгких: перегородки между альвеолами разрываются, несколько альвеол вместе сливаются в 1 воздушную полость, или кисту. Обнаруживают их при рентгенографическом обследовании. Разрыв такой буллы ведёт к спонтанному пневмотораксу, когда воздух выходит в плевральную полость. Буллы и пневмоторакс рассматривают как малые критерии синдрома Марфана.
Семейный анамнез
В случаях семейного анамнеза, когда есть подтверждённые случаи такой же болезни у родственников 1 линии (родители, дети, родные братья, сёстры), а также, когда при генетическом исследовании подтверждена мутация гена, ответственного за синтез фибриллина, достаточно 1 большого клинического критерия и вовлечения ещё 1 любой системы в патологический процесс. Достоверность диагноза повышается, если у 2 родственников выявлен большой критерий поражения скелета.
Если же в семье нет диагностированных случаев синдрома Марфана, необходимо наличие хотя бы 2 больших клинических критериев и поражение 3 системы в организме.
С какими состояниями следует дифференцировать
Есть много наследственных заболеваний, похожих по внешнему виду или характеру поражения внутренних органов:
- семейная эктопия хрусталика;
- семейный марфаноидный габитус (внешний вид);
- семейное расслоение аорты;
- врождённая контрактурная арахнодактилия (паучьи пальцы);
- семейная грудная аневризма аорты;
- семейный синдром пролапса митрального клапана.
Отличить их может только врач после детального обследования больного, от диагноза зависит дальнейшая тактика лечения.
Лечение
Как все генетически обусловленные заболевания, вылечить синдром Марфана нельзя. Можно корректировать возникающие нарушения.
Оперативное лечение показано при расширении аорты более 6 см в диаметре: укрепляют протезом поражённый участок сосуда.
При быстром прогрессировании патологии митрального клапана его заменяют на искусственный клапан. При вывихе или подвывихе хрусталика проводят операцию на глазах, фиксируют хрусталик в правильном положении.
Диета с большим содержанием магния положительно сказывается на течении заболевания, меньше поражаются сосуды. Препараты, уменьшающие сердечный выброс и артериальное давление (b -адреноблокаторы), уменьшают нагрузку на сердце и начальный отдел аорты. Они уменьшают риск внезапной смерти от сердечно-сосудистых осложнений. Т. к. поражён клапанный аппарат сердца, у больных повышенная склонность к развитию септического эндокардита. Любые вмешательства на сердце проводят с терапией антибиотиками для профилактики развития этого осложнения.
Пищевые добавки, содержащие магний, цинк, кальций, медь, витамин D3, гиалуроновую кислоту, викасол, могут несколько улучшить метаболизм и состояние соединительной ткани. С этой же целью назначают препараты аскорбиновой и янтарной кислоты, глюкозаминосульфаты и хондроитинсульфаты, карнитина хлорид, L-лизин, витамин Е.
Беременность
Риск проявления заболевания у потомства составляет 50%. Об этом следует предупреждать женщин до беременности. Кроме того, во время беременности значительно повышается нагрузка на сердце и аорту, особенно в последнем триместре, во время родов и в послеродовом периоде. Перестройка гормонального фона, кровообращения, увеличение объёма циркулирующей крови и артериального давления могут привести к расширению, расслоению, разрыву аорты и септическому эндокардиту.
Роды естественным путём разрешаются при диаметре аорты до 4 см по данным эхокардиографии. Причём для облегчения родов проводится эпидуральное обезболивание. Если диаметр аорты превышает 5,5 см, проводят операцию кесарево сечение.
В целом, синдром Марфана несёт определённую угрозу для жизни больного человека. Поэтому заболевание необходимо как можно раньше диагностировать. Больные должны соблюдать правильный режим и все рекомендации врачей, чтобы максимально снизить риск осложнений. Хирургическое лечение помогает исправить патологические изменения, возникшие в органах. Особенно бдительными должны быть люди, близкие родственники которых страдают подобным заболеванием. Женщинам с таким диагнозом рекомендуют учитывать риск при планировании беременности.
Что надо знать о синдроме Марфана, смотрите в тематическом видео:




Сообщить об опечатке
Текст, который будет отправлен нашим редакторам: